
Séquençage du code génétique avec le Machine Learning
En génomique, le code génétique peut être interprété comme le language de programmation du vivant, il permet aux différents éléments du corps de produire les molécules essentielles au fonctionnement physiologique. La compréhension de ce language est un élément important en taxonomie pour déterminer les différences entre les espèces, mais aussi en médecine pour déterminer le rôle de séquences indispensables. Ainsi, sur cette logique, il est pertinent de traiter ces séquences comme un language naturel (NLP) en utilisant la méthode des k-mers, bien connue en génomique. Ce projet permet de déterminer l'espèce dont est tirée la séquence d'ADN.
Processing et modélisation
Ce projet vise à classifier des séquences génétiques de différentes espèces (humain, chimpanzé, chien) en utilisant des techniques de traitement du langage naturel (NLP) et de Machine Learning. L'objectif est de transformer les séquences génétiques en mots k-mers, puis d'utiliser un modèle de classification pour prédire l'espèce à partir de ces mots.
Le pré-processing est une étape cruciale sur ce type de projet, car nous devons préparer les séquences génétiques à l'analyse. Pour cela, une fonction getKmers est définie dans l'objectif de convertir les chaines en mots de longueur fixe (les k-mers). Par défaut, ce sont des hexamères qui sont utilisés (longueur de 6 caractères). Cette transformation permet de représenter les séquences génétiques sous forme de mots, facilitant l'application des techniques de NLP.
Par la suite, cette fonction est appliquée aux données des trois espèces utilisées (humain, chimpanzé et chien) et les k-mers sont ensuite concaténés en une chaine de texte pour chaque séquence. Cette étape est indispensable pour assurer la compatibilité avec les algorithmes de Machine Learning.
Pour transformer les chaînes de texte en vecteurs de fréquence de mots, un CountVectorizer de la bibliothèque scikit-learn est employé. Cette étape permet de convertir les séquences de mots en une représentation numérique que les algorithmes de Machine Learning peuvent comprendre. Cette conversion est appliquée aux données de chaque espèce et les dimensions des vecteurs résultants sont vérifiées pour assurer que la transformation ait été effectuée correctement.
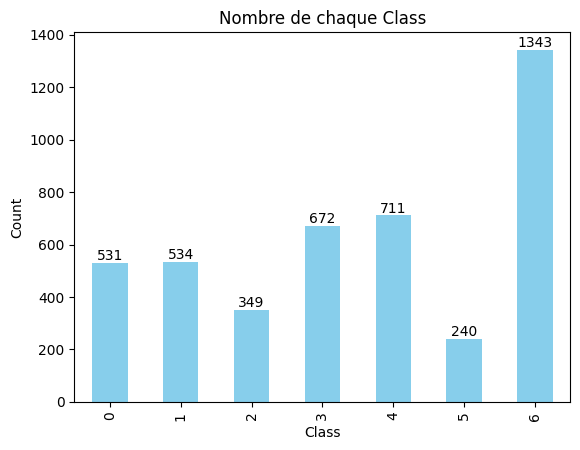
Histogramme du nombre de séquences de chaque classe
De manière classique, les données sont séparées en ensembles d'entrainement et de test grâce à la fonction train_test_split en réservant 20% des données pour le test et en utilisant les 80% restants pour l'entrainement. Ceci permet de garantir que le modèle est évalué de manière juste et objective.
Optimisation du modèle
Pour optimiser les hyperparamètres de notre modèle de classification, GridSearchCV est utilisé. Cette technique permet de rechercher les meilleurs hyperparamètres en utilisant la validation croisée. Une grille de paramètres pour le modèle MultinomialNB est définie et une recherche exhaustive est effectuée pour trouver la combinaison de paramètres qui maximise la précision du modèle. Cette étape est considérée comme essentielle pour améliorer la performance du modèle et garantir qu'il généralise bien aux nouvelles données.
Une fois les hyperparamètres optimisés, le modèle de classification MultinomialNB est entraîné sur les données d'entraînement. Ce modèle est bien adapté aux données textuelles et aux problèmes de classification en classes multiples. Le meilleur paramètre alpha trouvé lors de la recherche par grille est utilisé pour entraîner le modèle.
Pour évaluer la performance de notre modèle, plusieurs métriques de performance sont utilisées, notamment l'accuracy, la précision, le recall et le score F1. La matrice de confusion est également calculée pour visualiser les performances du modèle de manière détaillée. La matrice de confusion permet de voir combien de prédictions correctes et incorrectes ont été faites pour chaque classe et les résultats sont plus que satisfaisants avec une précision générale de 0.98.
Conclusion
Cette étude de cas démontre l'efficacité des techniques de NLP et de Machine Learning pour la classification des séquences génétiques. En transformant les séquences en mots k-mers et en utilisant un modèle de classification optimisé, nous pouvons obtenir des résultats précis et fiables. Cette approche peut être étendue à d'autres domaines de la bioinformatique et de la génomique, offrant de nouvelles perspectives pour l'analyse des données biologiques.